













Informatics Educational Institutions & Programs

| Buchwald-Hartwig amination | |
|---|---|
| Named after | Stephen L. Buchwald John F. Hartwig |
| Reaction type | Coupling reaction |
| Identifiers | |
| Organic Chemistry Portal | buchwald-hartwig-reaction |
| RSC ontology ID | RXNO:0000192 |
In organic chemistry, the Buchwald–Hartwig amination is a chemical reaction for the synthesis of carbon–nitrogen bonds via the palladium-catalyzed coupling reactions of amines with aryl halides.[1] Although Pd-catalyzed C–N couplings were reported as early as 1983, Stephen L. Buchwald and John F. Hartwig have been credited, whose publications between 1994 and the late 2000s established the scope of the transformation. The reaction's synthetic utility stems primarily from the shortcomings of typical methods (nucleophilic substitution, reductive amination, etc.) for the synthesis of aromatic C−N bonds, with most methods suffering from limited substrate scope and functional group tolerance.[2] The development of the Buchwald–Hartwig reaction allowed for the facile synthesis of aryl amines, replacing to an extent harsher methods (the Goldberg reaction, nucleophilic aromatic substitution, etc.) while significantly expanding the repertoire of possible C−N bond formations.[citation needed]

Over the course of its development, several 'generations' of catalyst systems have been developed, with each system allowing greater scope in terms of coupling partners and milder conditions, allowing virtually any amine to be coupled with a wide variety of aryl coupling partners.[citation needed] Because of the ubiquity of aryl C–N bonds in pharmaceuticals and natural products, the reaction has gained wide use in synthetic organic chemistry, with application in many total syntheses and the industrial preparation of numerous pharmaceuticals.
History
The first example of a palladium catalyzed C–N cross-coupling reaction was published in 1983 by Migita and coworkers and described a reaction between several aryl bromides and N,N-diethylamino-tributyltin using 1 mol% PdCl2[P(o-tolyl)3]2. Though several aryl bromides were tested, only electronically neutral, sterically unencumbered substrates gave good to excellent yields.[3]
-

Original precedent for Pd-catalyzed C–N coupling (Eq.2)

In 1984, Dale L. Boger and James S. Panek reported an example of Pd(0)-mediated C–N bond formation in the context of their work on the synthesis of lavendamycin which utilized stoichiometric Pd(PPh3)4. Attempts to render the reaction catalytic were unsuccessful.[4]
-

C–N coupling reaction in the total synthesis of lavendamycin (Eq.3)

These reports were virtually uncited for a decade. In February 1994, Hartwig reported a systematic study of the palladium compounds involved in the original Migita paper, concluding that the d10 complex Pd[P(o-Tolyl)3]2 was the active catalyst. Proposed was a catalytic cycle involving oxidative addition of the aryl bromide.[5]
-
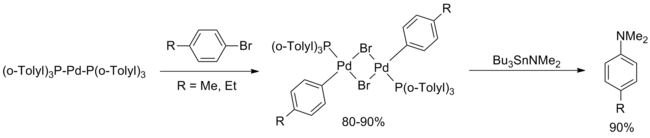
hartwig 1994 (Eq.4)

In May 1994, Buchwald published an extension of the Migita paper offering two major improvements over the original paper. First, transamination of Bu3SnNEt2 followed by argon purge to remove the volatile diethylamine allowed extension of the methodology to a variety of secondary amines (both cyclic and acyclic) and primary anilines. Secondly, the yield for electron rich and electron poor arenes was improved via minor modifications to the reaction procedure (higher catalyst loading, higher temperature, longer reaction time), although no ortho-substituted aryl groups were included in this publication.[6]
-

Buchwald 1994 publication (Eq.5)

In 1995, back to back studies from each lab showed that the couplings could be conducted with free amines in the presence of a bulky base (NaOtBu in the Buchwald publication, LiHMDS in the Hartwig publication), allowing for organotin-free coupling. Though these improved conditions proceeded at a faster rate, the substrate scope was limited almost entirely to secondary amines due to competitive hydrodehalogenation of the bromoarenes.[7][8] (See Mechanism below)
-

1995 Tin-free coupling conditions (Eq.6)

These results established the so-called "first generation" of Buchwald–Hartwig catalyst systems. The following years saw development of more sophisticated phosphine ligands that allowed extension to a larger variety of amines and aryl groups. Aryl iodides, chlorides, and triflates eventually became suitable substrates, and reactions run with weaker bases at room temperature were developed. These advances are detailed in the Scope section below, and the extension to more complex systems remains an active area of research.
Mechanism
The reaction mechanism for this reaction has been demonstrated to proceed through steps similar to those known for palladium catalyzed CC coupling reactions. Steps include oxidative addition of the aryl halide to a Pd(0) species, addition of the amine to the oxidative addition complex, deprotonation followed by reductive elimination. An unproductive side reaction can compete with reductive elimination wherein the amide undergoes beta hydride elimination to yield the hydrodehalogenated arene and an imine product.[9]
Throughout the development of the reaction the group sought to identify reaction intermediates through fundamental mechanistic studies. These studies have revealed a divergent reaction pathways depending on whether monodentate or chelating phosphine ligands are employed in the reaction, and a number of nuanced influences have been revealed (especially concerning the dialkylbiaryl phosphine ligands developed by Buchwald).
The catalytic cycle proceeds as follows:[10][11][12][13]
-
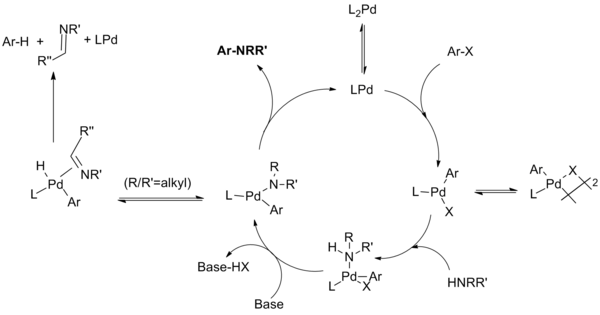
Catalytic cycle for monodentate phosphine ligand systems (Eq.7)

For monodentate ligand systems the monophosphine palladium (0) species is believed to form the palladium (II) species which is in equilibrium with the μ-halogen dimer. The stability of this dimer decreases in the order of X = I > Br > Cl, and is thought to be responsible for the slow reaction of aryl iodides with the first-generation catalyst system. Amine ligation followed by deprotonation by base produces the palladium amide. (Chelating systems have been shown to undergo these two steps in reverse order, with base complexation preceding amide formation.) This key intermediate reductively eliminates to produce the product and regenerate the catalyst. However, a side reaction can occur wherein β-hydride elimination followed by reductive elimination produces the hydrodehalogenated arene and the corresponding imine. Not shown are additional equilibria wherein various intermediates coordinate to additional phosphine ligands at various stages in the catalytic cycle.
For chelating ligands, the monophosphine palladium species is not formed; oxidative addition, amide formation and reductive elimination occur from L2Pd complexes. The Hartwig group found that "reductive elimination can occur from either a four-coordinate bisphosphine or three-coordinate monophosphine arylpalladium amido complex. Eliminations from the three-coordinate compounds are faster. Second, β-hydrogen elimination occurs from a three-coordinate intermediate. Therefore, β-hydrogen elimination occurs slowly from arylpalladium complexes containing chelating phosphines while reductive elimination can still occur from these four-coordinate species."[14]
Application
Because of the ubiquity of aryl C–N bonds in pharmaceuticals and natural products, the reaction has gained wide use in synthetic organic chemistry, with application in many total syntheses and the industrial preparation of numerous pharmaceuticals.[22] Industrial applications include α-arylation of carbonyl compounds (such as ketones, esters, amides, aldehydes) and nitriles.[23]
Scope
Although the scope of the Buchwald–Hartwig amination has been expanded to include a wide variety of aryl and amine coupling partners, the conditions required for any particular reactants are still largely substrate dependent. Various ligand systems have been developed, each with varying capabilities and limitations, and the choice of conditions requires consideration of the steric and electronic properties of both partners. Detailed below are the substrates and conditions for the major generations of ligand systems. (Not included herein are N-heterocyclic carbene ligands and ligands with wide bite angles such as Xantphos and Spanphos which also have been developed considerably.)[9]
First-generation catalyst system
The first generation (Pd[P(o-Tolyl)3]2) catalyst system was found to be effective for the coupling of both cyclic and acyclic secondary amines bearing both alkyl and aryl functionality (though not diarylamines) with a variety of aryl bromides. In general, these conditions were not able to couple primary amines due to competitive hydrodehalogenation of the arene.[7][8]
Aryl iodides were found to be suitable substrates for the intramolecular variant of this reaction,[8] and importantly, could be coupled intermolecularly only if dioxane was used in place of toluene as a solvent, albeit with modest yields.[24]

Bidentate phosphine ligands
The development of diphenylphosphinobinapthyl (BINAP) and diphenylphosphinoferrocene (DPPF) as ligands for the Buchwald–Hartwig amination provided the first reliable extension to primary amines and allowed efficient coupling of aryl iodides and triflates. (It is believed that the bidentate ligands prevent formation of the palladium iodide dimer after oxidative addition, speeding up the reaction.) These ligands typically produce the coupled products at higher rates and better yields than the first generation of catalysts. The initial reports of these ligands as catalysts were somewhat unexpected given the mechanistic evidence for monoligated complexes serving as the active catalysts in the first-generation system. In fact, the first examples from both labs were published in the same issue of JACS.[25][26][27]
-

Bidentate ligand examples (Eq.8)

The chelation from these ligands is thought to suppress β-hydride elimination by preventing an open coordination site. In fact, α-chiral amines were found not to racemize when chelating ligands were employed, in contrast to the first-generation catalyst system.[28]
-

Chiral retention by chelating phosphine ligands (Eq.9)

Sterically hindered ligands
Bulky tri- and di-alkyl phosphine ligands have been shown to be remarkably active catalysts, allowing the coupling of a wide range of amines (primary, secondary, electron withdrawn, heterocyclic, etc.) with aryl chlorides, bromides, iodides, and triflates. Additionally, reactions employing hydroxide, carbonate, and phosphate bases in place of the traditional alkoxide and silylamide bases have been developed. The Buchwald group has developed a wide range of dialkylbiaryl phosphine ligands, while the Hartwig group has focused on ferrocene-derived and trialkyl phosphine ligands.[29][30][31][32][33][34]
-

Bulky ligands in the Buchwald–Hartwig amination (Eq.10)

The dramatic increase in activity seen with these ligands is attributed to their propensity to sterically favor the monoligated palladium species at all stages of the catalytic cycle, dramatically increasing the rate of oxidative addition, amide formation, and reductive elimination. Several of these ligands also seem to enhance the rate of reductive elimination relative to β-hydride elimination via the electron donating arene-palladium interaction.[19][20]
Even electron withdrawn amines and heterocyclic substrates can be coupled under these conditions, despite their tendency to deactivate the palladium catalyst.[35][36]
-

Heteoaryl and amide substrates in the Buchwald–Hartwig amination (Eq.11)

Ammonia equivalents
Ammonia remains one of the most challenging coupling partners for Buchwald–Hartwig amination reactions, a problem attributed to its tight binding with palladium complexes. Several strategies have been developed to overcome this based on reagents that serve as ammonia equivalents. The use of a benzophenone imine or silylamide can overcome this limitation, with subsequent hydrolysis furnishing the primary aniline.[37][38][39]
-
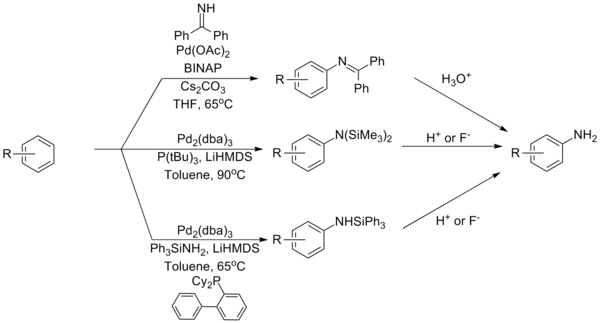
Ammonia equivalents in the Buchwald–Hartwig amination (Eq.12)

A catalyst system that can directly couple ammonia using a Josiphos-type ligand.[40]
Variations on C–N couplings: C–O, C–S, and C–C couplings
Under conditions similar to those employed for amination, alcohols can be coupled with aryl halides to produce the corresponding aryl ethers. This serves as a convenient replacement for harsher analogues of this process such as the Ullmann condensation.[41][42]
-

Aryl ether synthesis (Eq.13)

Thiols and thiophenols can be coupled with aryl halides under Buchwald-Hartwig-type conditions to produce the corresponding aryl thioethers. Furthermore, mercaptoesters have been employed as H2S-equivalents in order to generate the thiophenol from the corresponding aryl halide.[43]
Enolates and other similar carbon nucleophiles can also be coupled to produce α-aryl ketones, malonates, nitriles, etc. The scope of this transformation is similarly ligand-dependent and a number of systems have been developed.[44] Several enantioselective methods for this process have been developed.[45][46]
-

Enolate coupling as an extension of the Buchwald–Hartwig amination (Eq.14)

Several versions of the reaction employing complexes of copper and nickel rather than palladium have also been developed.[18]
References
- ^ Forero-Cortés, Paola A.; Haydl, Alexander M. (2 July 2019). "The 25th Anniversary of the Buchwald–Hartwig Amination: Development, Applications, and Outlook". Organic Process Research & Development. 23 (8): 1478–1483. doi:10.1021/acs.oprd.9b00161. S2CID 198366762.
- ^ Weygand, Conrad (1972). Hilgetag, G.; Martini, A. (eds.). Weygand/Hilgetag Preparative Organic Chemistry (4th ed.). New York: John Wiley & Sons, Inc. p. 461. ISBN 0471937495.
- ^ Kosugi, M.; Kameyama, M.; Migita, T. (1983), "Palladium-Catalyzed Aromatic Amination of Aryl Bromides With n,n-Di-Ethylamino-Tributyltin", Chemistry Letters, 12 (6): 927–928, doi:10.1246/cl.1983.927
- ^ Boger, D.L.; Panek, J.S. (1984), "Palladium(0)- mediated [beta]-carboline synthesis: Preparation of the CDE ring system of lavendamycin", Tetrahedron Letters, 25 (30): 3175–3178, doi:10.1016/S0040-4039(01)91001-9
- ^ Paul,F.; Patt, J.; Hartwig, J.F. (1994), "Palladium-catalyzed formation of carbon-nitrogen bonds. Reaction intermediates and catalyst improvements in the hetero cross-coupling of aryl halides and tin amides", J. Am. Chem. Soc., 116 (13): 5969–5970, doi:10.1021/ja00092a058
- ^ Guram, A.S.; Buchwald, S.L. (1994), "Palladium-Catalyzed Aromatic Aminations with in situ Generated Aminostannanes", J. Am. Chem. Soc., 116 (17): 7901–7902, doi:10.1021/ja00096a059
- ^ a b Louie,J.; Hartwig, J.F. (1995), "Palladium-catalyzed synthesis of arylamines from aryl halides. Mechanistic studies lead to coupling in the absence of tin reagents", Tetrahedron Letters, 36 (21): 3609–3612, doi:10.1016/0040-4039(95)00605-C
- ^ a b c Guram, A.S.; Rennels, R.A.; Buchwald, S.L. (1995), "A Simple Catalytic Method for the Conversion of Aryl Bromides to Arylamines", Angewandte Chemie International Edition, 34 (12): 1348–1350, doi:10.1002/anie.199513481
- ^ a b c Muci, A.R.; Buchwald, S.L. (2002), "Practical Palladium Catalysts for C–N and C–O Bond Formation", Topics in Curr. Chem., Topics in Current Chemistry, 219: 131–209, doi:10.1007/3-540-45313-x_5, ISBN 978-3-540-42175-7
- ^ Driver, M.S.; Hartwig, J.F. (1997), "Carbon−Nitrogen-Bond-Forming Reductive Elimination of Arylamines from Palladium(II) Phosphine Complexes", J. Am. Chem. Soc., 119 (35): 8232–8245, doi:10.1021/ja971057x
- ^ Hartwig, J.F.; Richards, S.; Barañano, D.; Paul, F. (1996), "Influences on the Relative Rates for C−N Bond-Forming Reductive Elimination and β-Hydrogen Elimination of Amides. A Case Study on the Origins of Competing Reduction in the Palladium-Catalyzed Amination of Aryl Halides", J. Am. Chem. Soc., 118 (15): 3626–3633, doi:10.1021/ja954121o
- ^ Driver, M.S.; Hartwig, J.F. (1995), "A Rare, Low-Valent Alkylamido Complex, a Diphenylamido Complex, and Their Reductive Elimination of Amines by Three-Coordinate Intermediates", J. Am. Chem. Soc., 117 (16): 4708–4709, doi:10.1021/ja00121a030
- ^ Widenhoefer, R.A.; Buchwald, S.L. (1996), "Halide and Amine Influence in the Equilibrium Formation of Palladium Tris(o-tolyl)phosphine Mono(amine) Complexes from Palladium Aryl Halide Dimers", Organometallics, 15 (12): 2755–2763, doi:10.1021/om9509608
- ^ a b Hartwig, J.F. (1999), "Approaches to catalyst discovery. New carbon-heteroatom and carbon-carbon bond formation", Pure Appl. Chem., 71 (8): 1416–1423, doi:10.1351/pac199971081417, S2CID 34700080
- ^ Hartwig, J.F. (1997), "Palladium-Catalyzed Amination of Aryl Halides: Mechanism and Rational Catalyst Design", Synlett, 1997 (4): 329–340, doi:10.1055/s-1997-789, S2CID 196704196
- ^ Hartwig, J.F. (1998), "Carbon-Heteroatom Bond-Forming Reductive Eliminations of Amines, Ethers, and Sulfides", Acc. Chem. Res., 31: 852–860, doi:10.1021/ar970282g
- ^ Wolfe, J.P.; Wagaw, S.; Marcoux, J.F.; Buchwald, S.L. (1998), "Rational Development of Practical Catalysts for Aromatic Carbon-Nitrogen Bond Formation", Acc. Chem. Res., 31: 805–818, doi:10.1021/ar9600650
- ^ a b Hartwig, J.F. (1998), "Transition Metal Catalyzed Synthesis of Arylamines and Aryl Ethers from Aryl Halides and Triflates: Scope and Mechanism", Angew. Chem. Int. Ed., 37 (15): 2046–2067, doi:10.1002/(sici)1521-3773(19980817)37:15<2046::aid-anie2046>3.0.co;2-l, PMID 29711045
- ^ a b Hartwig, J.F. (2008), "Evolution of a Fourth Generation Catalyst for the Amination and Thioetherification of Aryl Halides", Acc. Chem. Res., 41 (11): 1534–1544, doi:10.1021/ar800098p, PMC 2819174, PMID 18681463
- ^ a b Surry, D.S.; Buchwald, S.L. (2008), "Biaryl Phosphane Ligands in Palladium-Catalyzed Amination", Angew. Chem. Int. Ed., 47 (34): 6338–6361, doi:10.1002/anie.200800497, PMC 3517088, PMID 18663711
- ^ Surry, D.S.; Buchwald, S.L. (2011), "Dialkylbiaryl phosphines in Pd-catalyzed amination: a user's guide", Chem. Sci., 2 (1): 27–50, doi:10.1039/c0sc00331j, PMC 3306613, PMID 22432049
- ^ [15][16][14][9][17][18][19][20][21]
- ^ Thomas J. Colacot. The 2010 Nobel Prize in Chemistry: Palladium-Catalysed Cross-Coupling. Archived 2020-06-02 at the Wayback Machine Platinum Metals Rev., 2011, 55, (2) doi:10.1595/147106711X558301
- ^ Wolfe, J. P.; Buchwald, S. L. (1996), "Palladium-Catalyzed Amination of Aryl Iodides", J. Org. Chem., 61 (3): 1133–1135, doi:10.1021/jo951844h
- ^ Driver, M.S.; Hartwig, J.F. (1996), "A Second-Generation Catalyst for Aryl Halide Amination: Mixed Secondary Amines from Aryl Halides and Primary Amines Catalyzed by (DPPF)PdCl2", J. Am. Chem. Soc., 118 (30): 7217–7218, doi:10.1021/ja960937t
- ^ Wolfe, J.P.; Wagaw, S.; Buchwald, S.L. (1996), "An Improved Catalyst System for Aromatic Carbon-Nitrogen Bond Formation: The Possible Involvement of Bis(Phosphine) Palladium Complexes as Key Intermediates", J. Am. Chem. Soc., 118: 7215–7216, doi:10.1021/ja9608306
- ^ Louie, J.; Driver, M.S.; Hamann, B.C.; Hartwig, J.F. (1997), "Palladium-Catalyzed Amination of Aryl Triflates and Importance of Triflate Addition Rate", J. Org. Chem., 62 (5): 1268–1273, doi:10.1021/jo961930x
- ^ Wagaw, S.; Rennels, R.A.; Buchwald, S.L. (1997), "Palladium-Catalyzed Coupling of Optically Active Amines with Aryl Bromides", J. Am. Chem. Soc., 119 (36): 8451–8458, doi:10.1021/ja971583o
- ^ Old, D.W.; Wolfe, J.P.; Buchwald, S.L. (1998), "A Highly Active Catalyst for Palladium-Catalyzed Cross-Coupling Reactions: Room-Temperature Suzuki Couplings and Amination of Unactivated Aryl Chlorides", J. Am. Chem. Soc., 120 (37): 9722–9723, doi:10.1021/ja982250+
- ^ Wolfe, J.P.; Buchwald, S.L. (1999), "A Highly Active Catalyst for the Room-Temperature Amination and Suzuki Coupling of Aryl Chlorides", Angew. Chem. Int. Ed., 38 (16): 2413–2416, doi:10.1002/(sici)1521-3773(19990816)38:16<2413::aid-anie2413>3.0.co;2-h, PMID 10458806
- ^ Hamann, B.C.; Hartwig, J.F. (1998), "Sterically Hindered Chelating Alkyl Phosphines Provide Large Rate Accelerations in Palladium-Catalyzed Amination of Aryl Iodides, Bromides, and Chlorides, and the First Amination of Aryl Tosylates", J. Am. Chem. Soc., 120 (29): 7369–7370, doi:10.1021/ja981318i
- ^ Wolfe, J.P.; Tomori, H.; Sadighi, J.P.; Yin, J.; Buchwald, S.L. (2000), "Simple, Efficient Catalyst System for the Palladium-Catalyzed Amination of Aryl Chlorides, Bromides, and Triflates" (PDF), J. Org. Chem., 65 (4): 1158–1174, doi:10.1021/jo991699y, PMID 10814067
- ^ Stambuli, J.P.; Kuwano, R.; Hartwig, J.F. (2002), "Unparalleled Rates for the Activation of Aryl Chlorides and Bromides: Coupling with Amines and Boronic Acids in Minutes at Room Temperature", Angew. Chem. Int. Ed., 41 (24): 4746–4748, doi:10.1002/anie.200290036, PMID 12481346
- ^ Huang, X.; Anderson, K.W.; Zim, D.; Jiang, L.; Klapars, A.; Buchwald, S.L. (2003), "Expanding Pd-Catalyzed C–N Bond-Forming Processes: The First Amidation of Aryl Sulfonates, Aqueous Amination, and Complementarity with Cu-Catalyzed Reactions", J. Am. Chem. Soc., 125 (22): 6653–6655, doi:10.1021/ja035483w, PMID 12769573
- ^ Anderson, K.W.; Tundel, R.E.; Ikawa, T.; Altman, R.A.; Buchwald, S.L. (2006), "Monodentate Phosphines Provide Highly Active Catalysts for Pd-Catalyzed C–N Bond-Forming Reactions of Heteroaromatic Halides/Amines and (H)N-Heterocycles", Angew. Chem. Int. Ed., 45 (39): 6523–6527, doi:10.1002/anie.200601612, PMID 16955526
- ^ Ikawa, T.; Barder, T.E.; Biscoe, M.R.; Buchwald, S.L. (2007), "Pd-Catalyzed Amidations of Aryl Chlorides Using Monodentate Biaryl Phosphine Ligands: A Kinetic, Computational, and Synthetic Investigation", J. Am. Chem. Soc., 129 (43): 13001–13007, doi:10.1021/ja0717414, PMID 17918833
- ^ Wolfe, J.P.; Ahman, J.; Sadighi, J.P.; Singer, R.A.; Buchwald, S.L. (1997), "An Ammonia Equivalent for the Palladium-Catalyzed Amination of Aryl Halides and Triflates", Tetrahedron Lett., 38 (36): 6367–6370, doi:10.1016/S0040-4039(97)01465-2
- ^ Lee, S.; Jorgensen, M.; Hartwig, J.F. (2001), "Palladium-Catalyzed Synthesis of Arylamines from Aryl Halides and Lithium Bis(trimethylsilyl)amide as an Ammonia Equivalent", Org. Lett., 3 (17): 2729–2732, doi:10.1021/ol016333y, PMID 11506620
- ^ Huang, X.; Buchwald, S.L. (2001), "New Ammonia Equivalents for the Pd-Catalyzed Amination of Aryl Halides", Org. Lett., 3 (21): 3417–3419, doi:10.1021/ol0166808, PMID 11594848
- ^ Vo, G.D.; Hartwig, J.F. (2009), "Palladium-Catalyzed Coupling of Ammonia with Aryl Chlorides, Bromides, Iodides, and Sulfonates: A General Method for the Preparation of Primary Arylamines", J. Am. Chem. Soc., 131 (31): 11049–11061, doi:10.1021/ja903049z, PMC 2823124, PMID 19591470
- ^ Mann, G.; Incarvito, C.; Rheingold, A.L.; Hartwig, J.F. (1999), "Palladium-Catalyzed C–O Coupling Involving Unactivated Aryl Halides. Sterically Induced Reductive Elimination To Form the C–O Bond in Diaryl Ethers", J. Am. Chem. Soc., 121: 3224–3225, doi:10.1021/ja984321a
- ^ Torraca, K.E.; Huang, X.; Parrish, C.A.; Buchwald, S.L. (2001), "An Efficient Intermolecular Palladium-Catalyzed Synthesis of Aryl Ethers", J. Am. Chem. Soc., 123 (43): 10770–10771, doi:10.1021/ja016863p, PMID 11674023
- ^ Heesgaard Jepsen Tue (2011). "Synthesis of Functionalized Dibenzothiophenes - An Efficient Three-Step Approach Based on Pd-Catalyzed C–C and CS Bond Formations". European Journal of Organic Chemistry. 2011: 53–57. doi:10.1002/ejoc.201001393.
- ^ Culkin, D.A.; Hartwig, J.F. (2003), "Palladium-Catalyzed r-Arylation of Carbonyl Compounds and Nitriles", Acc. Chem. Res., 36 (4): 234–245, doi:10.1021/ar0201106, PMID 12693921
- ^ Hamada, T.; Chieffi, A.; Ahman, J.; Buchwald, S.L. (2002), "An Improved Catalyst for the Asymmetric Arylation of Ketone Enolates", J. Am. Chem. Soc., 124 (7): 1261–1268, doi:10.1021/ja011122+, PMID 11841295
- ^ Liao, X.; Weng, Z.; Hartwig, J.F. (2008), "Enantioselective r-Arylation of Ketones with Aryl Triflates Catalyzed by Difluorphos Complexes of Palladium and Nickel", J. Am. Chem. Soc., 130 (1): 195–200, doi:10.1021/ja074453g, PMC 2551326, PMID 18076166
External links
- Buchwald–Hartwig Coupling – Recent Literature
- Buchwald–Hartwig Chemistry Ian Mangion MacMillan Group Meeting July 30, 2002 Link
- Buchwald–Hartwig reaction Precious-Metal catalysts from Acros Organics for coupling reactions in organic synthesis Link
