













Informatics Educational Institutions & Programs


Heterogeneous gold catalysis refers to the use of elemental gold as a heterogeneous catalyst. As in most heterogeneous catalysis, the metal is typically supported on metal oxide. Furthermore, as seen in other heterogeneous catalysts, activity increases with a decreasing diameter of supported gold clusters. Several industrially relevant processes are also observed such as H2 activation, Water-gas shift reaction, and hydrogenation.[1][2][3] One or two gold-catalyzed reactions may have been commercialized.[4]
The high activity of supported gold clusters has been proposed to arise from a combination of structural changes, quantum-size effects and support effects that preferentially tune the electronic structure of gold[5] such that optimal binding of adsorbates during the catalytic cycle is enabled.[2][3][6] The selectivity and activity of gold nanoparticles can be finely tuned by varying the choice of support material, with e.g. titania (TiO2), hematite (α-Fe2O3), cobalt(II/III) oxide (Co3O4) and nickel(II) oxide (NiO) serving as the most effective support materials for facilitating the catalysis of CO combustion.[1] Besides enabling an optimal dispersion of the nanoclusters, the support materials have been suggested to promote catalysis by altering the size, shape, strain and charge state of the cluster.[3][7][8] A precise shape control of the deposited gold clusters has been shown to be important for optimizing the catalytic activity, with hemispherical, few atomic layers thick nanoparticles generally exhibiting the most desirable catalytic properties due to maximized number of high-energy edge and corner sites.[1][6][9]
Proposed applications
In the past, heterogeneous gold catalysts have found preliminary commercial applications for the industrial production of vinyl chloride (precursor to polyvinyl chloride or PVC) and methyl methacrylate.[4] Traditionally, PVC production uses mercury catalysts and leads to serious environmental concerns. China accounts for 50% of world's mercury emissions and 60% of China's mercury emission is caused by PVC production. Although gold catalysts are slightly expensive, overall production cost is affected by only ~1%. Therefore, green gold catalysis is considered valuable. The price fluctuation in gold has later led to cease the operations based on their use in catalytic converters. Very recently, there has been a lot of developments in gold catalysis for the synthesis of organic molecules including the C-C bond forming homocoupling or cross-coupling reactions and it has been speculated that some of these catalysts could find applications in various fields.[10]
CO oxidation
Gold can be a very active catalyst in oxidation of carbon monoxide (CO), i.e. the reaction of CO with molecular oxygen to produce carbon dioxide (CO2). Particles of 2 to 5 nm exhibit high catalytic activities. Supported gold clusters, thin films and nanoparticles are one to two orders of magnitude more active than atomically dispersed gold cations or unsupported metallic gold.[2]
Gold cations can be dispersed atomically on basic metal oxide supports such as MgO and La2O3. Monovalent and trivalent gold cations have been identified, the latter being more active but less stable than the former. The turnover frequency (TOF) of CO oxidation on these cationic gold catalysts is in the order of magnitude of 0.01 s−1, exhibiting the very high activation energy of 138 kJ/mol.[2]
Supported gold nanoclusters with a diameter < 2 nm are active to CO oxidation with turnover number (TOF) in the order of magnitude of 0.1 s−1. It has been observed that clusters with 8 to 100 atoms are catalytically active. The reason is that, on one hand, eight atoms are the minimum necessary to form a stable, discrete energy band structure, and on the other hand, d-band splitting decreases in clusters with more than 100 atoms, resembling the bulk electronic structure. The support has a substantial effect on the electronic structure of gold clusters. Metal hydroxide supports such as Be(OH)2, Mg(OH)2, and La(OH)3, with gold clusters of < 1.5 nm in diameter constitute highly active catalysts for CO oxidation at 200 K (-73 °C). By means of techniques such as HR-TEM and EXAFS, it has been proven that the activity of these catalysts is due exclusively to clusters with 13 atoms arranged in an icosahedron structure. Furthermore, the metal loading should exceed 10 wt% for the catalysts to be active.[2]
Gold nanoparticles in the size range of 2 to 5 nm catalyze CO oxidation with a TOF of about 1 s−1 at temperatures below 273 K (0 °C). The catalytic activity of nanoparticles is brought about in the absence of moisture when the support is semiconductive or reducible, e.g. TiO2, MnO2, Fe2O3, ZnO, ZrO2, or CeO2. However, when the support is insulating or non-reducible, e.g. Al2O3 and SiO2, a moisture level > 5000 ppm is required for activity at room temperature. In the case of powder catalysts prepared by wet methods, the surface OH− groups on the support provide sufficient aid as co-catalysts, so that no additional moisture is necessary. At temperatures above 333 K (60 °C), no water is needed at all.[2]
The apparent activation energy of CO oxidation on supported gold powder catalysts prepared by wet methods is 2-3 kJ/mol above 333 K (60 °C) and 26-34 kJ/mol below 333 K. These energies are low, compared to the values displayed by other noble metal catalysts (80-120 kJ/mol). The change in activation energy at 333 K can be ascribed to a change in reaction mechanism. This explanation has been supported experimentally. At 400 K (127 °C), the reaction rate per surface Au atom is not dependent on particle diameter, but the reaction rate per perimeter Au atom is directly proportional to particle diameter. This suggests that the mechanism above 333 K takes place on the gold surfaces. By contrast, at 300 K (27 °C), the reaction rate per surface Au atom is inversely proportional to particle diameter, while the rate per perimeter interface does not depend on particle size. Hence, CO oxidation occurs on the perimeter sites at room temperature. Further information on the reaction mechanism has been revealed by studying the dependency of the reaction rate on the partial pressures of the reactive species. Both at 300 K and 400 K, there is a first order rate dependency on CO partial pressure up to 4 Torr (533 Pa), above which the reaction is zero order. With respect to O2, the reaction is zero order above 10 Torr (54.7 kPa) at both 300 and 400 K. The order with respect to O2 at lower partial pressures is 1 at 300 K and 0.5 at 400 K. The shift towards zero order indicates that the catalyst's active sites are saturated with the species in question. Hence, a Langmuir-Hinshelwood mechanism has been proposed, in which CO adsorbed on gold surfaces reacts with O adsorbed at the edge sites of the gold nanoparticles.[2]
The need to use oxide supports, and more specifically reducible supports, is due to their ability to activate dioxygen. Gold nanoparticles supported on inert materials such as carbon or polymers have been proven inactive in CO oxidation. The aforementioned dependency of some catalysts on water or moisture also relates to oxygen activation. The ability of certain reducible oxides, such as MnO2, Co3O4, and NiO to activate oxygen in dry conditions (< 0.1 ppm H2O) can be ascribed to the formation of oxygen defects during pretreatment.[2]
Water gas shift
Water gas shift is the most widespread industrial process for the production of dihydrogen, H2. It involves the reaction of carbon monoxide and water (syngas) to form hydrogen and carbon dioxide as a byproduct. In many catalytic reaction schemes, one of the elementary reactions is the oxidation of CO with an adsorbed oxygen species. Gold catalysts have been proposed as an alternative for water gas shift at low temperatures, viz. < 523 K (250 °C). This technology is essential to the development of solid oxide fuel cells. Hematite has been found to be an appropriate catalyst support for this purpose. Furthermore, a bimetallic Au-Ru/Fe2O3 catalyst has been proven highly active and stable for low-temperature water gas shift. Titania and ceria have also been used as supports for effective catalysts. Unfortunately, Au/CeO2 is prone to deactivation caused by surface-bound carbonate or formate species.[12]
Although gold catalysts are active at room temperature to CO oxidation, the high amounts of water involved in water gas shift require higher temperatures. At such temperatures, gold is fully reduced to its metallic form. However, the activity of e.g. Au/CeO2 has been enhanced by CN− treatment, whereby metallic gold is leached, leaving behind highly active cations. According to DFT calculations, the presence of such Au cations on the catalyst is allowed by empty, localized nonbonding f states in CeO2. On the other hand, STEM studies of Au/CeO2 have revealed nanoparticles of 3 nm in diameter. Water gas shift has been proposed to occur at the interface of Au nanoparticles and the reduced CeO2 support.[12]
Epoxidations
Although the epoxidation of ethylene is routinely achieved in the industry with selectivities as high as 90% on Ag catalysts, most catalysts provided < 10% selectivity for propylene epoxidation. Using a gold catalyst supported on titanium silicate-1 (TS-1) molecular sieve, yields of 350 g/h per gram of gold were obtained at 473 K (200 °C). The reaction took place in the gas phase. Furthermore, using mesoporous titanosilicate supports (Ti-MCM-41 and Ti-MCM-48), gold catalysts provided > 90% selectivity at ~ 7% propylene conversion, 40% H2 efficiency, and 433 K (160 °C). The active species in these catalysts were identified to be hemispherical gold nano-crystals of less than 2 nm in diameter in intimate contact with the support.[12]
Alkene epoxidation has been demonstrated in absence of H2 reductant in the liquid phase. For example, using 1% Au/graphite, ~80% selectivities of cis-cyclooctene to cyclooctene oxide (analogous to cyclohexene oxide) were obtained at 7-8% conversion, 353 K (80 °C), and 3 MPa O2 in absence of hydrogen or solvent.[12] Other liquid-phase selective oxidations have been achieved with saturated hydrocarbons. For instance, cyclohexane has been converted to cyclohexanone and cyclohexanol with a combined selectivity of ~100% on gold catalysts. Product selectivities can be tuned in liquid phase reactions by the presence or absence of solvent and by the nature of the latter, viz. water, polar, or nonpolar. With gold catalysts, the catalyst's support has less influence on reactions in the liquid phase than on reactions in the gas phase.[13]
Selective hydrogenations
Typical hydrogenation catalysts are based on metals from the 8, 9, and 10 groups, such as Ni, Ru, Pd, and Pt. By comparison, gold has a poor catalytic activity for hydrogenation.[14] This low activity is caused by the difficulty of dihydrogen activation on gold. While hydrogen dissociates on Pd and Pt without an energy barrier, dissociation on Au(111) has an energy barrier of ~1.3 eV, according to DFT calculations. These calculations agree with experimental studies, in which hydrogen dissociation was not observed on gold (111) or (110) terraces, nor on (331) steps. No dissociation was observed on these surfaces either at room temperature or at 473 K (200 °C). However, the rate of hydrogen activation increases for Au nanoparticles.[2] Notwithstanding its poor activity, nano-sized gold immobilized in various supports has been found to provide a good selectivity in hydrogenation reactions.[14]

One of the early studies (1966) of hydrogenation on supported, highly dispersed gold was performed with 1-butene and cyclohexene in the gas phase at 383 K (110 °C). The reaction rate was found to be first order with trespect to alkene pressure and second order with respect to chemisorbed hydrogen. In later works, it was shown that gold-catalyzed hydrogenation can be highly sensitive to Au loading (hence to particle size) and to the nature of the support. For example, 1-pentene hydrogenation occurred optimally on 0.04 wt% Au/SiO2, but not at all on Au/γ-Al2O3.[12] By contrast, the hydrogenation of 1,3-butadiene to 1-butene was shown to be relatively insensitive to Au particle size in a study with a series of Au/Al2O3 catalysts prepared by different methods. With all the tested catalysts, conversion was ~100% and selectivity, < 60%.[14] Concerning reaction mechanisms, in a study of propylene hydrogenation on Au/SiO2, reaction rates were determined using D2 and H2. Because the reaction with deuterium was substantially slower, it was suggested that the rate-determining step in alkene hydrogenation was the cleavage of the H-H bond. Lastly, ethylene hydrogenation was studied on Au/MgO at atmospheric pressure and 353 K (80 °C) with EXAFS, XANES and IR spectroscopy, suggesting that the active species might be Au+3 and the reaction intermediate, an ethylgold species.[12]
Gold catalysts are especially selective in the hydrogenation of α,β-insaturated aldehydes, i.e. aldehydes containing a C=C double bond on the carbon adjacent to the carbonyl. Gold catalysts are able to hydrogenate only the carbonyl group, so that the aldehyde is transformed to the corresponding alcohol, while leaving the C=C double bond untouched. In the hydrogenation of crotonaldehyde to crotyl alcohol, 80% selectivity was attained at 5-10% conversion and 523 K (250 °C) on Au/ZrO2 and Au/ZnO. The selectivity increased along with Au particle size in the range of ~2 to ~5 nm. Other instances of this reaction include acrolein, citral, benzal acetone, and pent-3-en-2-one. The activity and selectivity of gold catalysts for this reaction has been linked to the morphology of the nanoparticles, which in turn is influenced by the support. For example, round particles tend to form on TiO2, while ZnO promotes particles with clear facets, as observed by TEM. Because the round morphology provides a higher relative amount of low-coordinated metal surface sites, the higher activity observerd with Au/TiO2 compared to Au/ZnO is explained. Finally, a bimetallic Au-In/ZnO catalyst has been observed to improve the selectivity towards the hydrogenation of the carbonyl in acrolein. It was observed in HRTEM images that indium thin films decorate some of the facets of the gold nanoparticle. The promoting effect on selectivity might result from the fact that only the Au sites that promote side-reactions are decorated by In.[12]
A strategy that in many reactions has succeeded at improving gold's catalytic activity without impairing its selectivity is to synthesize bimetallic Pd-Au or Pt-Au catalysts. For the hydrogenation of 1,3-butadiene to butenes, model surfaces of Au(111), Pd-Au(111), Pd-Au(110), and Pd(111) were studied with LEED, AES, and LEIS. A selectivity of ~100% was achieved on Pd70Au30(111) and it was suggested that Au might promote the desorption of the product during the reaction. A second instance is the hydrogenation of p-chloronitrobenzene to p-chloroaniline, in which selectivity suffers with typical hydrogenation catalysts due to the parallel hydrodechlorination to aniline. However, Pd-Au/Al2O3 (Au/Pd ≥20) has been proven thrice as active as the pure Au catalyst, while being ~100% selective to p-chloroaniline. In a mechanistic study of hydrogenation of nitrobenzenes with Pt-Au/TiO2, the dissociation of H2 was identified as rate-controlling, hence the incorporation of Pt, an efficient hydrogenation metal, highly improved catalytic activity. Dihydrogen dissociated on Pt and the nitroaromatic compound was activated on the Au-TiO2 interface. Finally, hydrogenation was enabled by the spillover of activated H surface species from Pt to the Au surface.[14][15]
Theoretical background
Bulk metallic gold is known to be inert, exhibiting a surface reactivity at room temperature only towards a few substances such as formic acid and sulphur-containing compounds, e.g. H2S and thiols.[1] Within heterogeneous catalysis, reactants adsorb onto the surface of the catalyst thus forming activated intermediates. However, if the adsorption is weak such as in the case of bulk gold, a sufficient perturbation of the reactant electronic structure does not occur and catalysis is hindered (Sabatier's principle). When gold is deposited as nanosized clusters of less than 5 nm onto metal oxide supports, a markedly increased interaction with adsorbates is observed, thereby resulting in surprising catalytic activities. Evidently, nano-scaling and dispersing gold on metal oxide substrates makes gold less noble by tuning its electronic structure, but the precise mechanisms underlying this phenomenon are as of yet uncertain and hence widely studied.[3][13][16]
It is generally known that decreasing the size of metallic particles in some dimension to the nanometer scale will yield clusters with a significantly more discrete electronic band structure in comparison with the bulk material.[9] This is an example of a quantum-size effect and has been previously correlated with an increased reactivity enabling nanoparticles to bind gas phase molecules more strongly. In the case of TiO2-supported gold nanoparticles, Valden et al.[2] observed the opening of a band gap of approximately 0.2-0.6 eV in the gold electronic structure as the thickness of the deposited particles was decreased below three atomic layers. The two-layer thick supported gold clusters were also shown to be exceptionally active for CO combustion, based on which it was concluded that quantum-size effects inducing a metal-insulator transition play a key role in enhancing the catalytic properties of gold. However, decreasing the size further to a single atomic layer and a diameter of less than 3 nm was reported to again decrease the activity. This has later been explained by a destabilization of clusters composed of very few atoms, resulting in too strong bonding of adsorbates and thus poisoning of the catalyst.[3][8]
The properties of the metal d-band are central for describing the origin of catalytic activity based on electronic effects.[17] According to the d-band model of heterogeneous catalysis, substrate-adsorbate bonds are formed as the discrete energy levels of the adsorbate molecule interacts with the metal d-band, thus forming bonding and antibonding orbitals. The strength of the formed bond depends on the position of the d-band center such that a d-band closer to the Fermi level () will result in stronger interaction. The d-band center of bulk gold is located far below , which qualitatively explains the observed weak binding of adsorbates as both the bonding and antibonding orbitals formed upon adsorption will be occupied, resulting in no net bonding.[17] However, as the size of gold clusters is decreased below 5 nm, it has been shown that the d-band center of gold shifts to energies closer to the Fermi level, such that the as formed antibonding orbital will be pushed to an energy above , hence reducing its filling.[18][19] In addition to a shift in the d-band center of gold clusters, the size-dependency of the d-band width as well as the spin-orbit splitting has been studied from the viewpoint of catalytic activity.[20] As the size of the gold clusters is decreased below 150 atoms (diameter ca. 2.5 nm), rapid drops in both values occur. This can be attributed to d-band narrowing due to the decreased number of hybridizing valence states of small clusters as well as to the increased ratio of high-energy edge atoms with low coordination to the total number of Au atoms. The effect of the decreased spin-orbit splitting as well as the narrower distribution of d-band states on the catalytic properties of gold clusters cannot be understood via simple qualitative arguments as in the case of the d-band center model. Nevertheless, the observed trends provide further evidence that a significant perturbation of the Au electronic structure occurs upon nanoscaling, which is likely to play a key role in the enhancement of the catalytic properties of gold nanoparticles.


A central structural argument explaining the high activity of metal oxide supported gold clusters is based on the concept of periphery sites formed at the junction between the gold cluster and the substrate.[1][2] In the case of CO oxidation, it has been hypothesized that CO adsorbs onto the edges and corners of the gold clusters, while the activation of oxygen occurs at the peripheral sites. The high activity of edge and corner sites towards adsorption can be understood by considering the high coordinative unsaturation of these atoms in comparison with terrace atoms. The low degree of coordination increases the surface energy of corner and edge sites, hence making them more active towards binding adsorbates. This is further coupled with the local shift of the d-band center of the unsaturated Au atoms towards energies closer to the Fermi level, which in accordance with the d-band model results in increased substrate-adsorbate interaction and lowering of the adsorption-dissociation energy barriers.[17][20] Lopez et al.[18] calculated the adsorption energy of CO and O2 on the Au(111) terrace on which the Au-atoms have a coordination number of 9 as well as on an Au10 cluster where the most reactive sites have a coordination of 4. They observed that the bond strengths are in general increased by as much as 1 eV, indicating a significant activation towards CO oxidation if one assumes that the activation barriers of surface reactions scale linearly with the adsorption energies (Brønsted-Evans-Polanyi principle). The observation that hemispherical two-layer gold clusters with a diameter of a few nanometers are most active for CO oxidation is well in line with the assumption that edge and corner atoms serve as the active sites, since for clusters of this shape and size the ratio of edge atoms to the total number of atoms is indeed maximized.[9]
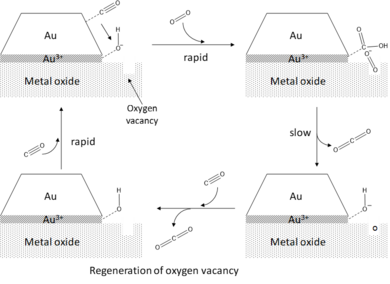
The preferential activation of O2 at the perimeter sites is an example of a support effect that promotes the catalytic activity of gold nanoparticles. Besides enabling a proper dispersion of the deposited particles and hence a high surface-to-volume ratio, the metal oxide support also directly perturbs the electronic structure of the deposited gold clusters via various mechanisms, including strain-inducing and charge transfer. For gold deposited on magnesia (MgO), a charge transfer from singly charged oxygen vacancies (F-centers) at the MgO surface to the Au cluster has been observed.[8] This charge transfer induces a local perturbation in the electronic structure of the gold clusters at the perimeter sites, enabling the formation of resonance states as the antibonding orbital of oxygen interacts with the metal d-band. As the antibonding orbital is occupied, the O-O bond is significantly weakened and stretched, i.e. activated. In gas-phase model studies, the formation of activated super-oxo species O2- is found to correlate with the size-dependent electronic properties of the clusters.[21][22] The activation of O2 at the perimeter sites is also observed for defect-free surfaces and neutral gold clusters, but to a significantly smaller extent. The activity enhancing effect of charge transfer from the substrate to gold has also been reported by Chen and Goodman[7] in the case of a gold bilayer supported on ultrathin TiO2 on Mo(112). In addition to charge transfer between the substrate and the gold nanoparticles, the support material has been observed to increase the catalytic activity of gold by inducing strain as a consequence of lattice mismatch.[9] The induced strains especially affect the Au atoms close to the substrate-cluster interface, resulting in a shift of the local d-band center towards energies closer to the Fermi level. This corroborates the periphery hypothesis and the creation of catalytically active bifunctional sites at the cluster-support interface.[3] Furthermore, the support-cluster interaction directly influences the size and shape of the deposited gold nanoparticles. In the case of weak interaction, less active 3D clusters are formed, whereas if the interaction is stronger more active 2D few-layer structures are formed. This illustrates the ability to fine-tune the catalytic activity of gold clusters via varying the support material as well as the underlying metal upon which the substrate has been grown.[8][19]

Finally, it has been observed that the catalytic activity of supported gold clusters towards CO oxidation is further enhanced by the presence of water.[2] Invoking the periphery hypothesis, water promotes the activation of O2 by co-adsorption onto the perimeter sites where it reacts with O2 to form adsorbed hydroxyl (OH*) and hydroperoxo (OOH*) species. The reaction of these intermediates with adsorbed CO is very rapid, and results in the efficient formation of CO2 with concomitant recovery of the water molecule.[8]
See also
References
- ^ a b c d e Haruta, Masatake (1997). "Size- and support-dependency in the catalysis of gold". Catalysis Today. 36 (1): 153–166. doi:10.1016/s0920-5861(96)00208-8.
- ^ a b c d e f g h i j k l Haruta, Masatake (2011-10-04). "Spiers Memorial Lecture : Role of perimeter interfaces in catalysis by gold nanoparticles". Faraday Discussions. 152: 11–32, discussion 99–120. Bibcode:2011FaDi..152...11H. doi:10.1039/c1fd00107h. ISSN 1364-5498. PMID 22455036.
- ^ a b c d e f van Santen, Rutger Anthony; Neurock, Matthew (2006). Molecular heterogeneous catalysis. A conceptual and computational approach. Weinheim, Germany: Wiley-VCH. pp. 53–60. ISBN 978-3-527-29662-0.
- ^ a b Ciriminna, Rosaria; Falletta, Ermelinda; Della Pina, Cristina; Teles, Joaquim Henrique; Pagliaro, Mario (2016). "Industrial Applications of Gold Catalysis". Angewandte Chemie International Edition. 55 (46): 1433–7851. doi:10.1002/anie.201604656. hdl:2434/463818. PMID 27624999. S2CID 28730917.
- ^ P. Chatterjee; H. Wang; J. S. Manzano; U. Kanbur; A. D. Sadow; I. I. Slowing (2022). "Surface ligands enhance the catalytic activity of supported Au nanoparticles for the aerobic α-oxidation of amines to amides". Catal. Sci. Technol. 12 (6): 1922–1933. doi:10.1039/D1CY02121D. S2CID 246575960.
- ^ a b Valden, M.; Lai, X.; Goodman, D. W. (1998-09-11). "Onset of Catalytic Activity of Gold Clusters on Titania with the Appearance of Nonmetallic Properties". Science. 281 (5383): 1647–1650. Bibcode:1998Sci...281.1647V. doi:10.1126/science.281.5383.1647. ISSN 0036-8075. PMID 9733505.
- ^ a b Chen, M. S.; Goodman, D. W. (2004-10-08). "The Structure of Catalytically Active Gold on Titania". Science. 306 (5694): 252–255. Bibcode:2004Sci...306..252C. doi:10.1126/science.1102420. ISSN 0036-8075. PMID 15331772. S2CID 19323471.
- ^ a b c d e Landman, Uzi; Yoon, Bokwon; Zhang, Chun; Heiz, Ueli; Arenz, Matthias (2007-06-01). "Factors in gold nanocatalysis: oxidation of CO in the non-scalable size regime". Topics in Catalysis. 44 (1–2): 145–158. CiteSeerX 10.1.1.459.9120. doi:10.1007/s11244-007-0288-6. ISSN 1022-5528. S2CID 17560286.
- ^ a b c d Mavrikakis, M.; Stoltze, P.; Nørskov, J. K. (2000-02-01). "Making gold less noble". Catalysis Letters. 64 (2–4): 101–106. doi:10.1023/A:1019028229377. ISSN 1011-372X. S2CID 96863829.
- ^ Nijamudheen, A.; Datta, Ayan (2020). "Gold‐Catalyzed Cross‐Coupling Reactions: An Overview of Design Strategies, Mechanistic Studies, and Applications". Chemistry: A European Journal. 26 (7): 1442–1487. doi:10.1002/chem.201903377. PMID 31657487. S2CID 204947412.
- ^ Haruta, Masatake (2011). "Spiers Memorial Lecture : Role of perimeter interfaces in catalysis by gold nanoparticles". Faraday Discussions. 152: 11–32, discussion 99–120. Bibcode:2011FaDi..152...11H. doi:10.1039/c1fd00107h. ISSN 1359-6640. PMID 22455036.
- ^ a b c d e f g Hashmi, A. Stephen K.; Hutchings, Graham J. (2006-12-04). "Gold Catalysis". Angewandte Chemie International Edition. 45 (47): 7896–7936. doi:10.1002/anie.200602454. ISSN 1521-3773. PMID 17131371.
- ^ a b Haruta, Masatake (October 2005). "Gold rush". Nature. 437 (7062): 1098–1099. doi:10.1038/4371098a. ISSN 1476-4687. PMID 16237427. S2CID 4347776.
- ^ a b c d Zhang, Yan; Cui, Xinjiang; Shi, Feng; Deng, Youquan (2012-04-11). "Nano-Gold Catalysis in Fine Chemical Synthesis". Chemical Reviews. 112 (4): 2467–2505. doi:10.1021/cr200260m. ISSN 0009-2665. PMID 22112240.
- ^ a b Serna, Pedro; Concepción, Patricia; Corma, Avelino (2009-07-01). "Design of highly active and chemoselective bimetallic gold–platinum hydrogenation catalysts through kinetic and isotopic studies". Journal of Catalysis. 265 (1): 19–25. doi:10.1016/j.jcat.2009.04.004. ISSN 0021-9517.
- ^ Turner, Mark; Golovko, Vladimir B.; Vaughan, Owain P. H.; Abdulkin, Pavel; Berenguer-Murcia, Angel; Tikhov, Mintcho S.; Johnson, Brian F. G.; Lambert, Richard M. (August 2008). "Selective oxidation with dioxygen by gold nanoparticle catalysts derived from 55-atom clusters". Nature. 454 (7207): 981–983. Bibcode:2008Natur.454..981T. doi:10.1038/nature07194. ISSN 1476-4687. PMID 18719586. S2CID 4355469.
- ^ a b c Hammer, B.; Norskov, J. K. (July 1995). "Why gold is the noblest of all the metals". Nature. 376 (6537): 238–240. Bibcode:1995Natur.376..238H. doi:10.1038/376238a0. ISSN 1476-4687. S2CID 4334587.
- ^ a b Lopez, N (2004). "On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation". Journal of Catalysis. 223 (1): 232–235. doi:10.1016/j.jcat.2004.01.001.
- ^ a b Jiang, T.; Mowbray, D. J.; Dobrin, S.; Falsig, H.; Hvolbæk, B.; Bligaard, T.; Nørskov, J. K. (2009-06-18). "Trends in CO Oxidation Rates for Metal Nanoparticles and Close-Packed, Stepped, and Kinked Surfaces". The Journal of Physical Chemistry C. 113 (24): 10548–10553. doi:10.1021/jp811185g. ISSN 1932-7447.
- ^ a b Visikovskiy, Anton; Matsumoto, Hisashi; Mitsuhara, Kei; Nakada, Toshitaka; Akita, Tomoki; Kido, Yoshiaki (2011). "Electronic d-band properties of gold nanoclusters grown on amorphous carbon". Physical Review B. 83 (16): 165428. Bibcode:2011PhRvB..83p5428V. doi:10.1103/physrevb.83.165428.
- ^ Woodham, Alex P.; Meijer, Gerard; Fielicke, André (2012-04-27). "Activation of Molecular Oxygen by Anionic Gold Clusters". Angewandte Chemie International Edition. 51 (18): 4444–4447. doi:10.1002/anie.201108958. hdl:11858/00-001M-0000-000F-895B-1. ISSN 1433-7851.
- ^ Woodham, Alex P.; Fielicke, André (2013), Mingos, D. Michael P. (ed.), "Gold Clusters in the Gas Phase", Gold Clusters, Colloids and Nanoparticles I, vol. 161, Cham: Springer International Publishing, pp. 243–278, doi:10.1007/430_2013_136, hdl:11858/00-001M-0000-0014-691E-E, ISBN 978-3-319-07847-2, retrieved 2023-10-23
