













Informatics Educational Institutions & Programs

A tetrahedral intermediate is a reaction intermediate in which the bond arrangement around an initially double-bonded carbon atom has been transformed from trigonal to tetrahedral.[1] Tetrahedral intermediates result from nucleophilic addition to a carbonyl group. The stability of tetrahedral intermediate depends on the ability of the groups attached to the new tetrahedral carbon atom to leave with the negative charge. Tetrahedral intermediates are very significant in organic syntheses and biological systems as a key intermediate in esterification, transesterification, ester hydrolysis, formation and hydrolysis of amides and peptides, hydride reductions, and other chemical reactions.
History
One of the earliest accounts of the tetrahedral intermediate came from Rainer Ludwig Claisen in 1887.[2] In the reaction of benzyl benzoate with sodium methoxide, and methyl benzoate with sodium benzyloxide, he observed a white precipitate which under acidic conditions yields benzyl benzoate, methyl benzoate, methanol, and benzyl alcohol. He named the likely common intermediate “additionelle Verbindung.”

Victor Grignard assumed the existence of unstable tetrahedral intermediate in 1901, while investigating the reaction of esters with organomagnesium reagents.[3]
The first evidence for tetrahedral intermediates in the substitution reactions of carboxylic derivatives was provided by Myron L. Bender in 1951.[4] He labeled carboxylic acid derivatives with oxygen isotope O18 and reacted these derivatives with water to make labeled carboxylic acids. At the end of the reaction he found that the remaining starting material had a decreased proportion of labeled oxygen, which is consistent with the existence of the tetrahedral intermediate.
Reaction mechanism

The nucleophilic attack on the carbonyl group proceeds via the Bürgi-Dunitz trajectory. The angle between the line of nucleophilic attack and the C-O bond is greater than 90˚ due to a better orbital overlap between the HOMO of the nucleophile and the π* LUMO of the C-O double bond.
Structure of tetrahedral intermediates
General features
Although the tetrahedral intermediates are usually transient intermediates, many compounds of this general structures are known. The reactions of aldehydes, ketones, and their derivatives frequently have a detectable tetrahedral intermediate, while for the reactions of derivatives of carboxylic acids this is not the case. At the oxidation level of carboxylic acid derivatives, the groups such as OR, OAr, NR2, or Cl are conjugated with the carbonyl group, which means that addition to the carbonyl group is thermodynamically less favored than addition to corresponding aldehyde or ketone. Stable tetrahedral intermediates of carboxylic acid derivatives do exist and they usually possess at least one of the following four structural features:
- polycyclic structures (e.g. tetrodotoxin)[5]
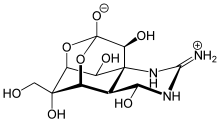
Tetrodotoxin - compounds with a strong electron-withdrawing group attached to the acyl carbon (e.g. N,N-dimethyltrifluoroacetamide)[6]
- compounds with donor groups that are poorly conjugated with the potential carbonyl group (e.g. cyclol)[7]
- compounds with sulfur atoms bonded to the anomeric centre (e.g., S-acylated-1,8-naphthalenedithiol)[8]

These compounds were used to study the kinetics of tetrahedral intermediate decomposition into its respective carbonyl species, and to measure the IR, UV, and NMR spectra of the tetrahedral adduct.
X-ray crystal structure determination
The first X-ray crystal structures of tetrahedral intermediates were obtained in 1973 from bovine trypsin crystallized with bovine pancreatic trypsin inhibitor,[9] and in 1974 from porcine trypsin crystallized with soybean trypsin inhibitor.[10] In both cases the tetrahedral intermediate is stabilized in the active sites of enzymes, which have evolved to stabilize the transition state of peptide hydrolysis.
Some insight into the structure of tetrahedral intermediate can be obtained from the crystal structure of N-brosylmitomycin A, crystallized in 1967.[11] The tetrahedral carbon C17 forms a 136.54 pm bond with O3, which is shorter than C8-O3 bond (142.31 pm). In contrast, C17-N2 bond (149.06 pm) is longer than N1-C1 bond (148.75 pm) and N1-C11 bond (147.85 pm) due to donation of O3 lone pair into σ* orbital of C17-N2. This model however is forced into tetracyclic sceleton, and tetrahedral O3 is methylated which makes it a poor model overall.

The more recent x-ray crystal structure of 1-aza-3,5,7-trimethyladamantan-2-one is a good model for cationic tetrahedral intermediate.[12] The C1-N1 bond is rather long [155.2(4) pm], and C1-O1(2) bonds are shortened [138.2(4) pm]. The protonated nitrogen atom N1 is a great amine leaving group.

In 2002 David Evans et al. observed a very stable neutral tetrahedral intermediate in the reaction of N-acylpyrroles with organometallic compounds, followed by protonation with ammonium chloride producing a carbinol.[13] The C1-N1 bond [147.84(14) pm] is longer than the usual Csp3-Npyrrole bond which range from 141.2-145.8 pm. In contrast, the C1-O1 bond [141.15(13) pm] is shorter than the average Csp3-OH bond which is about 143.2 pm. The elongated C1-N1, and shortened C1-O1 bonds are explained with an anomeric effect resulting from the interaction of the oxygen lone pairs with the σ*C-N orbital. Similarly, an interaction of an oxygen lone pair with σ*C-C orbital should be responsible for the lengthened C1-C2 bond [152.75(15) pm] compared to the average Csp2-Csp2 bonds which are 151.3 pm. Also, the C1-C11 bond [152.16(17) pm] is slightly shorter than the average Csp3-Csp3 bond which is around 153.0 pm.

Stability of tetrahedral intermediates
Acetals and hemiacetals
Hemiacetals and acetals are essentially tetrahedral intermediates. They form when nucleophiles add to a carbonyl group, but unlike tetrahedral intermediates they can be very stable and used as protective groups in synthetic chemistry. A very well known reaction occurs when acetaldehyde is dissolved in methanol, producing a hemiacetal. Most hemiacetals are unstable with respect to their parent aldehydes and alcohols. For example, the equilibrium constant for reaction of acetaldehyde with simple alcohols is about 0.5, where the equilibrium constant is defined as K = [hemiacetal]/[aldehyde][alcohol]. Hemiacetals of ketones (sometimes called hemiketals) are even less stable than those of aldehydes. However, cyclic hemiacetals and hemiacetals bearing electron withdrawing groups are stable. Electron-withdrawing groups attached to the carbonyl atom shift the equilibrium constant toward the hemiacetal. They increase the polarization of the carbonyl group, which already has a positively polarized carbonyl carbon, and make it even more prone to attack by a nucleophile. The chart below shows the extent of hydration of some carbonyl compounds. Hexafluoroacetone is probably the most hydrated carbonyl compound possible. Formaldehyde reacts with water so readily because its substituents are very small- a purely steric effect.[14][15]

Cyclopropanones- three-membered ring ketones- are also hydrated to a significant extent. Since three-membered rings are very strained (bond angles forced to be 60˚), sp3 hybridization is more favorable than sp2 hybridization. For the sp3 hybridized hydrate the bonds have to be distorted by about 49˚, while for the sp2 hybridized ketone the bond angle distortion is about 60˚. So the addition to the carbonyl group allows some of the strain inherent in the small ring to be released, which is why cyclopropanone and cyclobutanone are very reactive electrophiles. For larger rings, where the bond angles are not as distorted, the stability of the hemiacetals is due to entropy and the proximity of the nucleophile to the carbonyl group. Formation of an acyclic acetal involves a decrease in entropy because two molecules are consumed for every one produced. In contrast, the formation of cyclic hemiacetals involves a single molecule reacting with itself, making the reaction more favorable. Another way to understand the stability of cyclic hemiacetals is to look at the equilibrium constant as the ratio of the forward and backward reaction rate. For a cyclic hemiacetal the reaction is intramolecular so the nucleophile is always held close to the carbonyl group ready to attack, so the forward rate of reaction is much higher than the backward rate. Many biologically relevant sugars, such as glucose, are cyclic hemiacetals.

In the presence of acid, hemiacetals can undergo an elimination reaction, losing the oxygen atom that once belonged to the parent aldehyde’s carbonyl group. These oxonium ions are powerful electrophiles, and react rapidly with a second molecule of alcohol to form new, stable compounds, called acetals. The whole mechanism of acetal formation from hemiacetal is drawn below.

Acetals, as already pointed out, are stable tetrahedral intermediates so they can be used as protective groups in organic synthesis. Acetals are stable under basic conditions, so they can be used to protect ketones from a base. The acetal group is hydrolyzed under acidic conditions. An example with a dioxolane protecting group is given below.

Weinreb amides
Weinreb amides are N-methoxy-N-methylcarboxylic acid amides.[16] Weinreb amides are reacted with organometallic compounds to give, on protonation, ketones (see Weinreb ketone synthesis). It is generally accepted that the high yields of ketones are due to the high stability of the chelated five-membered ring intermediate. Quantum mechanical calculations have shown that the tetrahedral adduct is formed easily and it is fairly stable, in agreement with the experimental results.[17] The very facile reaction of Weinreb amides with organolithium and Grignard reagents results from the chelate stabilization in the tetrahedral adduct and, more importantly, the transition state leading to the adduct. The tetrahedral adducts are shown below.

Applications in biomedicine
Drug design
A solvated ligand that binds the protein of interest is likely to exist as an equilibrium mixture of several conformers. Likewise the solvated protein also exists as several conformers in equilibrium. Formation of protein-ligand complex includes displacement of the solvent molecules that occupy the binding site of the ligand, to produce a solvated complex. Because this necessarily means that the interaction is entropically disfavored, highly favorable enthalpic contacts between the protein and the ligand must compensate for the entropic loss. The design of new ligands is usually based on the modification of known ligands for the target proteins. Proteases are enzymes that catalyze hydrolysis of a peptide bond. These proteins have evolved to recognize and bind the transition state of peptide hydrolysis reaction which is a tetrahedral intermediate. Therefore, the main protease inhibitors are tetrahedral intermediate mimics having an alcohol or a phosphate group. Examples are saquinavir, ritonavir, pepstatin, etc.[18]
Enzymatic activity
Stabilization of tetrahedral intermediates inside of the enzyme active site has been investigated using tetrahedral intermediate mimics. The specific binding forces involved in stabilizing the transition state have been describe crystallographycally. In the mammalian serine proteases, trypsin and chymotrypsin, two peptide NH groups of the polypeptide backbone form the so-called oxyanion hole by donating hydrogen bonds to the negatively charged oxygen atom of the tetrahedral intermediate.[19] A simple diagram describing the interaction is shown below.

References
- ^ "IUPAC Gold Book definition". doi:10.1351/goldbook.T06289.
{{cite journal}}: Cite journal requires|journal=(help) - ^ Claisen, L. (1887). "Ueber die Einwirkung von Natriumalkylaten auf Benzaldehyd". Chem. Ber. 20 (1): 646–650. doi:10.1002/cber.188702001148.
- ^ Grignard, V. (1901). "Mixed organomagnesium combinations and their application in acid, alcohol, and hydrocarbon synthesis". Ann. Chim. Phys. 24: 433–490.
- ^ Bender, M. L. (1951). "Oxygen Exchange as Evidence for the Existence of an Intermediate in Ester Hydrolysis". J. Am. Chem. Soc. 73 (4): 1626–1629. doi:10.1021/ja01148a063.
- ^ Woodward, R. B.; Gougoutas, J. Z. (1964). "The Structure of Tetrodotoxin". J. Am. Chem. Soc. 86 (22): 5030. doi:10.1021/ja01076a076.
- ^ Gideon, Fraenkel; Watson Debra (1975). "Alkoxide adduct of an amide. Mean lifetime of an intimate ion pair". J. Am. Chem. Soc. 97 (1): 231–232. doi:10.1021/ja00834a063.
- ^ Cerrini, S.; Fedeli W.; Mazza F. (1971). "X-Ray crystallographic proof of a cyclol structure in a tripeptide". Chem. Commun. (24): 1607–1608. doi:10.1039/C29710001607.
- ^ Tagaki, M.; Ishahara R.; Matsudu T. (1977). "Mono S-Acylated 1,8-Naphthalenedithiol. Isolation and Characterization of Tetrahedral Intermediate in the Intramolecular Acyl Transfer Reaction". Bull. Chem. Soc. Jpn. 50 (8): 2193–2194. doi:10.1246/bcsj.50.2193.
- ^ Ruhlmann, A.; Kukla D.; Schwager P.; Bartels K.; Huber R. (1973). "Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor. Crystal structure determination and stereochemistry of the contact region". J. Mol. Biol. 77 (3): 417–436. doi:10.1016/0022-2836(73)90448-8. PMID 4737866.
- ^ Sweet, R.M.; Wright H.T.; Clothia C.H.; Blow D.M. (1974). "Crystal structure of the complex of porcine trypsin with soybean trypsin inhibitor (Kunitz) at 2.6 Å resolution". Biochemistry. 13 (20): 4212–4228. doi:10.1021/bi00717a024. PMID 4472048.
- ^ Tulinsky, A.; Van den Hende J.H. (1967). "The crystal and molecular structure of N-brosylmitomycin A". J. Am. Chem. Soc. 89 (12): 2905–2911. doi:10.1021/ja00988a018. PMID 6043811.
- ^ Kirby, A. J.; Komarov I.V.; Feeder N. (1998). "Spontaneous, Millisecond Formation of a Twisted Amide from the Amino Acid, and the Crystal Structure of a Tetrahedral Intermediate". J. Am. Chem. Soc. 120 (28): 7101–7102. doi:10.1021/ja980700s.
- ^ Evans, D. A.; G. Borg; K. A. Scheidt (2002). "Remarkably Stable Tetrahedral Intermediates: Carbinols from Nucleophilic Additions to N–Acylpyrroles". Angewandte Chemie. 114 (17): 3320–23. doi:10.1002/1521-3757(20020902)114:17<3320::aid-ange3320>3.0.co;2-u.
- ^ Bell, R. P. (1966). "The reversible hydration of carbonyl compounds". Adv. Phys. Org. Chem. Advances in Physical Organic Chemistry. 4 (1): 1–29. doi:10.1016/S0065-3160(08)60351-2. ISBN 9780120335046.
- ^ Clayden J.; Greeves N.; Warren S. & Wothers P. (2001). Organic Chemistry. Oxford University Press.
- ^ Nahm, S.; Weinreb, S. M. (1981). "N-methoxy-N-methylamides as effective acylating agents". Tetrahedron Lett. 22 (39): 3815–18. doi:10.1016/s0040-4039(01)91316-4.
- ^ Adler, M.; Adler S.; Boche G. (2005). "Tetrahedral intermediates in reactions of carboxylic acid derivatives with nucleophiles". J. Phys. Org. Chem. 18 (3): 193–209. doi:10.1002/poc.807.
- ^ Babine, R. E.; Bender S. L. (1997). "Molecular Recognition of Protein−Ligand Complexes: Applications to Drug Design". Chem. Rev. 97 (5): 1359–1472. doi:10.1021/cr960370z. PMID 11851455.
- ^ Bryan, P.; Pantoliano M. W.; Quill S. G.; Hsiao H. Y.; Poulos T. (1986). "Site-directed mutagenesis and the role of the oxyanion hole in subtilisin". Proc. Natl. Acad. Sci. USA. 83 (11): 3743–5. Bibcode:1986PNAS...83.3743B. doi:10.1073/pnas.83.11.3743. PMC 323599. PMID 3520553.
